Research project
Background
The combustion of conventional fuels, including kerosene, gasoline, and diesel, has been widely recognized as a significant source of pollutants that contribute to various environmental issues and pose significant risks to human health. A promising alternative fuel that has gained attention is ammonia. As an efficient hydrogen carrier, ammonia exhibits a high octane number, indicating its ability to resist knocking in practical engines. However, with high ignition temperature, low flame speed, and high NOx emissions, ammonia was limited from being widely used. To overcome these limitations, ammonia is often blended with other fuels with higher reactivity, such as hydrogen, natural gas, dimethyl ether (DME), gasoline, and diesel, etc. The feasibility of ammonia/gasoline and ammonia/biofuel blend combustion has been proven in practical engines under various conditions. This blending approach allows for the optimization of combustion characteristics and facilitates the practical implementation of ammonia as a viable alternative fuel source.
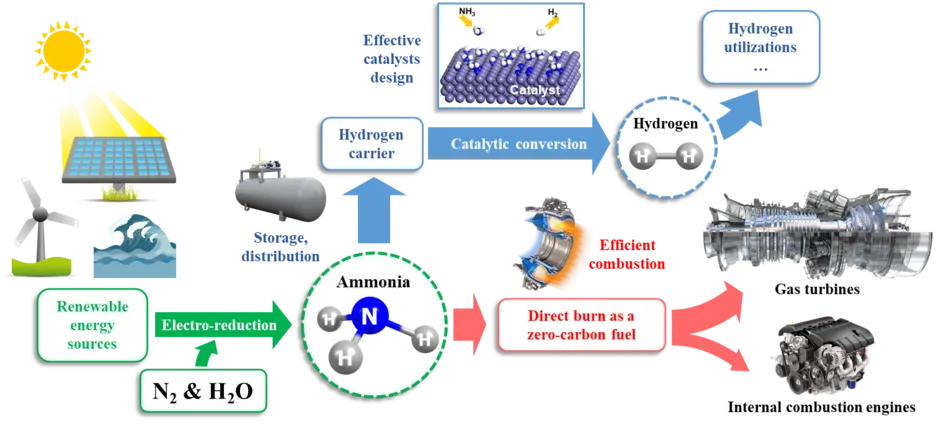
Research Content
A systematic understanding of the combustion behavior demands chemical knowledge. Chemical kinetic modeling is an important tool in the analysis of many combustion systems. When describing the combustion process of ammonia/fuel blends, hydrogen atom abstraction from species in fuels by ṄH2 radical is of significant importance. Therefore, a series of investigations about hydrogen atom abstraction from alkanes and esters by ṄH2 radical have been studied from high-level quantum ab initio to combustion model application.
(1) H-atom Abstraction from Alkanes by ṄH2 Radicals
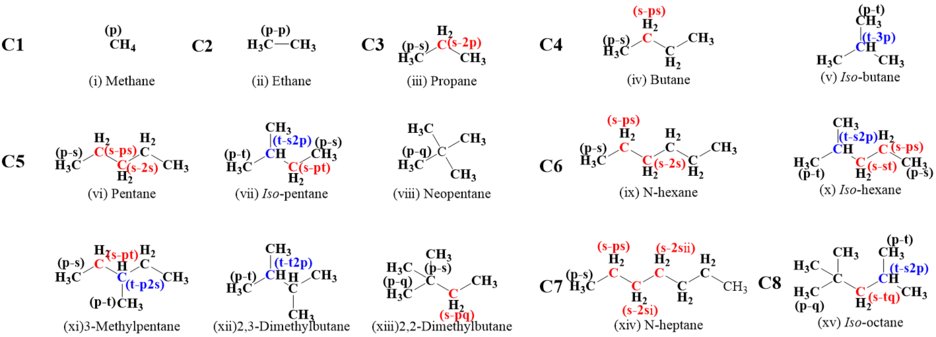
A systematic, theoretical chemical kinetic investigation of hydrogen atom abstraction by ṄH2 from C1-C6 alkanes, n-heptane and iso-octane has been performed. The geometry optimizations, frequencies, zero-point energies of all species related to the title reactions were calculated at the M06-2X/6-311++G(d,p) level of theory. One-dimensional hindered rotor treatment has also been performed at the same level of theory to obtain the potential energy surface for the torsional modes. Single-point energy calculations were obtained by using the QCISD(T) method combining with the cc-pVDZ and cc-pVTZ basis sets with correlations from MP2 with cc-pVDZ, cc-pVTZ, and cc-pVQZ basis sets. The high-pressure limiting rate constants of 41 reactions in the temperature range 298.15–2000 K were calculated using the Master Equation System Solver (MESS) with conventional transition state theory and comparison with existing available literature data were conducted. The generally good agreement was gained between our results with the available theoretical and experimental data. Rate rules were derived by analyzing the numerical relationship between the rate constants of various reaction site types. Additionally, the average hydrogen abstraction rate constants for primary, secondary, and tertiary type sites were reported on a per hydrogen atom basis. The obtained data and rules are valuable for the development of combustion kinetic models for ammonia and blending fuels.
(2) H-atom Abstraction from Esters by ṄH2 Radicals
Esters, a key component of biomass, can be added to gasoline to enhance performance or stability and to conventional gasoline or diesel fuel to improve lubricity, reduce emissions, and enhance combustion efficiency. Their cross-reactions with ṄH2 are essential for determining the reactivity of ammonia/biofuel or ammonia/diesel blends.
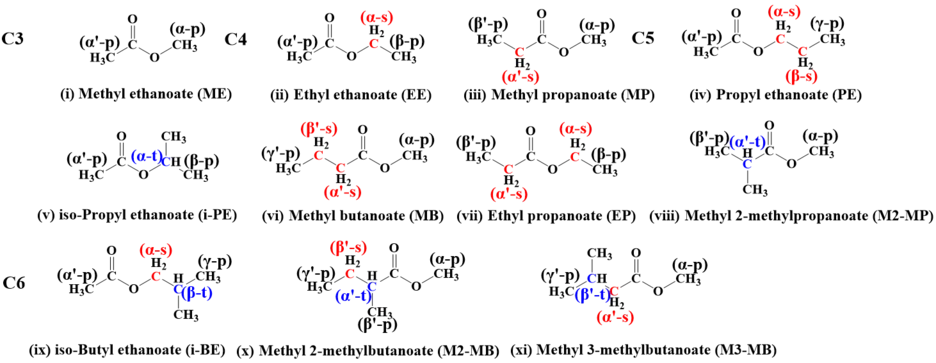
In this work, we have applied high-level ab initio calculations to conduct a series of theoretical chemical kinetic studies on H-atom abstraction from eleven alky esters of CnH2n+1COOCH3 (n =1–4), CH3COOCmH2m+1 (m=1–4), and C2H5COOC2H5 by ṄH2 radicals. The rate constants of 39 reaction channels over a temperature range of 500–2000 K were calculated. The impact of functional groups on the reactivity of H-atom abstraction by ṄH2 radical was systematically investigated by comparing with our previous work on alkanes, alcohols, and ethers with ṄH2 radical. Meanwhile, the recommended rate constant for different sites at esters was reported on a per hydrogen atom basis using the calculated average rate constant. These data could be directly used to construct kinetic mechanisms for larger esters and ammonia chemistry.
(3) Kinetics of H-atom abstractions from NH3 and H2NȮ by CH3Ȯ2: possible carbon-nitrogen interaction reactions
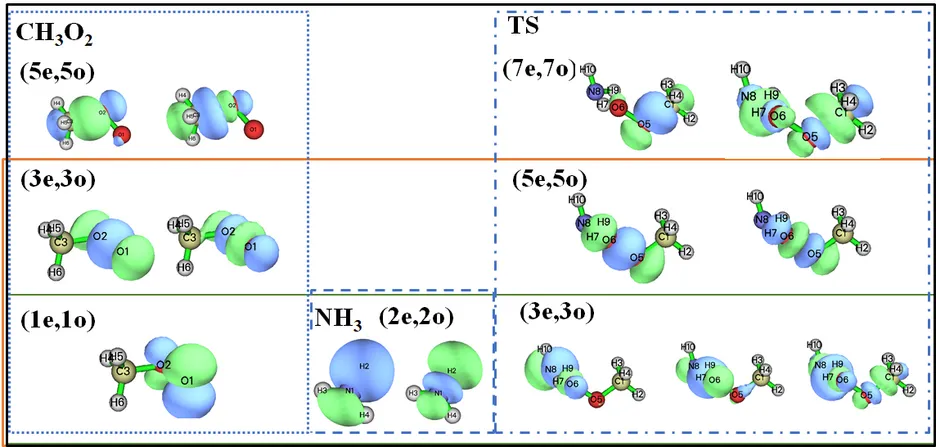
Understanding the low-temperature chemistry of ammonia (NH3) and its interaction with hydrocarbon chemistry poses a significant challenge in combustion kinetics. This is particularly relevant with the growing interest in novel combustion concepts and the increasing use of NH3 as an energy carrier. Carbon-nitrogen (C-N) interaction reactions play an important role in the combustion characteristic of NH3 blends with methane (CH4). In this work, the kinetics of two possible C-N interaction reactions, H-atom abstractions from NH3 and aminoxyl radical (H2NȮ) by methylperoxy radical (CH3Ȯ2), is investigated. These reaction pathways show some sensitivity to the low-temperature ignitions of NH3/CH4. However, they have not been considered in any combustion kinetic models so far. Due to the intrinsic multi-reference nature of their transition states, the energy barriers for the two reactions were determined using CASPT2/aug-cc-pVTZ method with active spaces of (7e,7o), and CASPT2/cc-pVDZ method with active spaces of (14e,12o), respectively, based on optimized geometries and rovibrational properties obtained at M06-2X/6-311++G(d,p) level of theory. Rate constants for the two reactions in the temperature range 298.15–2000 K are calculated using transition state theory. By incorporating these two reaction pathways with our calculated rate constants into an NH3/CH4 kinetic model, the predicted low-temperature ignition delay times (IDT) of NH3/CH4 mixtures become noticeably faster.
Publications
[1] Jingwu Sun, Yuxiang Zhu, Alexander A. Konnov, Chong-Wen Zhou*. From electronic structure to combustion model application for acrolein chemistry part I: Acrolein + H reactions and related chemistry[J]. Combustion and Flame (2021): 111825.
[2] Jingwu Sun, Yuxiang Zhu, JinTao Chen, Alexander A. Konnov, Ting Li, Lijun Yang, Chong-Wen Zhou*. "From electronic structure to combustion model application for acrolein chemistry Part Ⅱ: Acrolein+ HȮ2 reactions and the development of acrolein sub-mechanism." Combustion and Flame 251 (2023): 112321.
[3] Jingwu Sun, Zhaolin Fu, Hetian Zhu, Henry Zhiping Tao, Dongsheng Wen, Chong-Wen Zhou*. Theoretical Kinetic Study of Key Reactions Between Ammonia and Fuel Molecules, Part I: Hydrogen Atom Abstraction from Alkanes by ṄH2. Combustion and Flame 261 (2024): 113264.